

The Second Half of AI in Biomedicine
We are at the second half of AI in Biomedicine.

AI-driven biomedicine has spent a decade and tens of billions of dollars on drug discovery: finding the molecule, folding the protein, designing the antibody, and proving it does something to a cell or an animal. Call that the first half. We have effectively turned this from a scarcity problem into an abundance problem. Now the chokepoint sits downstream, in clinical development. Call that the second half, where “can we make this compound” turns into “what will it do inside an actual human body.” We don’t have an AI for that second question yet.
Everyone funded the first half of the pipeline
The scoreboard for the first half looks remarkable. AlphaFold cracked a fifty-year-old structural biology problem and won Demis Hassabis and John Jumper a Nobel Prize. By 2023, Boston Consulting Group’s analysis for the Wellcome Trust had tracked more than $18 billion invested across roughly 200 “AI-first” biotechs over the prior decade, with 60 percent of that capital concentrated in the top 20 companies. By 2024, BCG’s clinical follow-up counted at least 75 AI-discovered drugs and vaccines that had entered human trials. Isomorphic Labs signed research deals worth close to $3 billion with Eli Lilly and Novartis in January 2024, then in May 2026 raised a $2.1 billion Series B, the second-largest biotech financing round on record after Altos Labs, without having disclosed a single drug candidate of its own. Insilico Medicine’s rentosertib, an AI-proposed target for lung fibrosis paired with an AI-generated molecule, entered Phase III trials in July 2026, about as far as an AI-discovered drug has ever gotten.
Discovery isn’t the bottleneck anymore
Nobody in the field needs convincing that discovery got faster and cheaper. What that money hasn’t bought is an approved drug. BenevolentAI’s lead candidate, a topical eczema treatment built on the company’s own target-identification platform, missed its efficacy endpoints in a Phase 2a trial in 2023; the company cut 180 jobs within weeks, and its shares had fallen nearly 80% within the year. Even rentosertib, the furthest-along case anyone can point to, still has to clear Phase III, manufacturing, and regulatory review before it treats a single patient commercially. AI-driven discovery has flooded the pipeline with high-potential candidates. It has not yet bought a drug.
This is what you’d expect once the tractable half of a problem gets solved and the harder half doesn’t move. BCG’s own clinical analysis found that AI-discovered molecules clear Phase I at 80% to 90%, far above the 40% to 65% historical baseline, because Phase I mostly tests safety and pharmacokinetics, exactly the properties a model can be trained to optimize for. Phase II, where a drug meets a real efficacy signal in real patients for the first time, erases that advantage: AI-discovered molecules succeed at roughly the same rate as everything else, and that rate is the worst attrition point in the entire pipeline. Independent phase-transition data from MIT’s Wong, Siah, and Lo, built from more than 400,000 clinical trial records, puts Phase II success rates in the roughly 30% range, the lowest of any phase. Run the full gauntlet and the odds barely move: BCG’s own numbers put the overall probability that a molecule reaches approval at 5% to 10% for a human-discovered candidate and 9% to 18% for an AI-discovered one. Better molecule selection helps. It does not touch the part of the funnel that is actually killing candidates.

Why the second half is different, not just harder
Dismissing clinical trials as merely hard ignores the specific, quantifiable mechanisms causing them to fail. This 2019 modeling study estimated that the false discovery rate built into preclinical target identification runs above 92%: of all plausible protein-disease pairings that clear cell and animal screening, only about 1 in 200 is real. A late-stage failure for lack of efficacy is, in that light, an expensive way of discovering that the target never mattered, a fact only discoverable once a real trial finally puts the question to a human being. But “lack of efficacy” is itself a label sitting on top of a second, quieter failure: a trial is designed and enrolled before anyone knows which patients the drug will actually help. Response rates to drugs that work, not failed ones, vary enormously by patient. Statins alone range from 30% to 70% effectiveness across users, depending on genetic and metabolic factors. A trial protocol locks in its inclusion and exclusion criteria at the design stage, before dosing starts, using whatever biomarkers or clinical features are already known to correlate with response. When no such biomarker exists, which is the common case, the trial enrolls a genetically and physiologically heterogeneous population and measures one average effect across all of it. If a meaningful share of enrolled patients were never going to respond regardless of dose, the trial reads as a failed drug rather than a badly targeted one. The FDA’s own guidance on enrichment strategies exists to fix exactly this, by enrolling only the patients likely to respond, but it only works when a predictive marker for response is already known going in. Most of the time, it isn’t. Trial design cannot select for responders it has no way to identify in advance, which is a prediction problem, not a design flaw the next protocol can fix.
This has already sparked a race to build AI models that predict real efficacy. Look at where the papers and the money are actually pointed, though, and most of that effort sits one layer below the patient: at the cell. A large coalition spanning Stanford, the Chan Zuckerberg Initiative, and Genentech formalized the idea of a “virtual cell” in 2024: a foundation model that predicts how a cell’s state changes under a perturbation, rather than merely describing the cell as it sits today. Models like GEARS and CPA already do a version of this, predicting a cell’s transcriptional response to genetic or chemical perturbations it was never trained on, and the Arc Institute now runs a standing Virtual Cell Challenge to benchmark exactly that capability across labs. The 2025 results showed submitted models still failing to consistently beat a naive baseline. Even the easiest version of this problem, predicting one cell type’s response at one fixed timepoint, hasn’t been cracked yet.
A patient isn’t a monoculture of one cell type responding in isolation, and that’s not a scale problem, it’s a category problem. The same drug hits a diabetic liver differently than a healthy one, interacts with whatever else the patient already takes, and plays out against a specific immune history and genetic background, all of it compounding over months. Perturbation screens capture one transcriptional readout at one fixed point after one intervention. Scaling that up, more cell types, bigger perturbation libraries, doesn’t close the gap to a months-long therapeutic arc. It produces a sharper picture of the wrong unit of analysis.
Today, we already treat “predict the reaction of an intervention on the system” as the right question to ask about a cell or a tissue. Yet, we have simply not gotten around to asking the same question about a patient.
There’s also a reason nobody built the patient-level version first, and it isn’t neglect. Perturb-seq can generate thousands of clean, labeled, independent perturbation-response pairs in a single experiment, exactly the kind of data modern machine learning is built to exploit. A patient’s response to treatment is none of that: it’s one long, right-censored, confounded trajectory per person, shaped by which treatment a clinician chose to give them in the first place. Cell data is the kind of dataset a model can be benchmarked against by Tuesday. Patient data isn’t just longitudinal tracking; it’s a map of heterogeneous treatment effects. The field went where the benchmarks were easy to build, not where the stakes were highest, and BCG’s own report all but says so: it notes that time and cost savings in discovery are the easy win, while the change that would actually matter, a real lift in clinical probability of success, is the one nobody has delivered yet.
The missing patient world model
State it plainly: biomedicine needs a model that, given a patient’s current state (history-fused rather than a single snapshot) and a specific action taken on it, predicts what that patient’s state becomes next.
That quantity already has a name in another field. World model is what reinforcement learning calls a model of a system’s transition dynamics under an action, as distinct from a model of what the system merely looks like right now. David Ha and Jürgen Schmidhuber’s 2018 paper built an agent that learned a compressed model of a video game’s dynamics and let a controller plan by imagining rollouts inside that learned model rather than acting in the real environment at every step. That built on Richard Sutton’s Dyna architecture for planning inside a learned environment model, which itself traces back to Kenneth Craik’s 1943 proposal that minds work by building small-scale models of reality to anticipate what happens next. The term has drifted well past game-playing agents since, but the core meaning hasn’t: a world model predicts what happens under an action. It doesn’t just describe what is.
Fei-Fei Li’s recent taxonomy is useful here even though she was writing about the physical world, because it pins down exactly what that requires. In general, this field splits the concept into three functional parts: a renderer, which reconstructs what a scene looks like; a simulator, which predicts how that scene evolves under an action; and a planner, which uses the simulated future to choose the next action. Run biomedicine through the same lens and the gap becomes obvious. Genomics, pathology, imaging, wearables, and electronic health records already give us increasingly rich renderers of a patient’s biological state. Clinicians are the planners; they make treatment decisions constantly. What’s missing is the simulator: something that takes a patient’s rendered state, applies an action (this drug, this dose, this schedule), and predicts the actual trajectory that follows, not a mouse’s trajectory, not a trial population’s average, but this patient’s. That missing simulator is the linchpin of clinical development. It answers the one question that the second half is all about: what happens to this patient if we do this.
One concrete attempt
Our own effort at this, which we call StandardModel, starts from what a patient actually looks like in the data: not one biological scale but several at once, genomics, pathology, imaging, and the whole-organism trajectory recorded in clinical records over months and years. The reason to fuse them isn’t to see more of any single one. It’s that a lot of what determines outcome only shows up in how the scales interact. DNA methylation and miRNA expression each carry genuinely distinct prognostic signal rather than redundant copies of the same one, and integrated multi-omic models have been shown to outperform single-omics approaches precisely because a single measurement can’t capture the heterogeneity that actually drives variation in treatment response. That logic compounds further up the chain: a genetic variant’s effect on a patient’s trajectory is often contingent on organ function, comorbidity, and treatment history, none of which the genome alone encodes. A model trained on one layer can be arbitrarily good at that layer and still miss the outcome, because the signal that matters lives in the coupling between layers, not in any one of them.
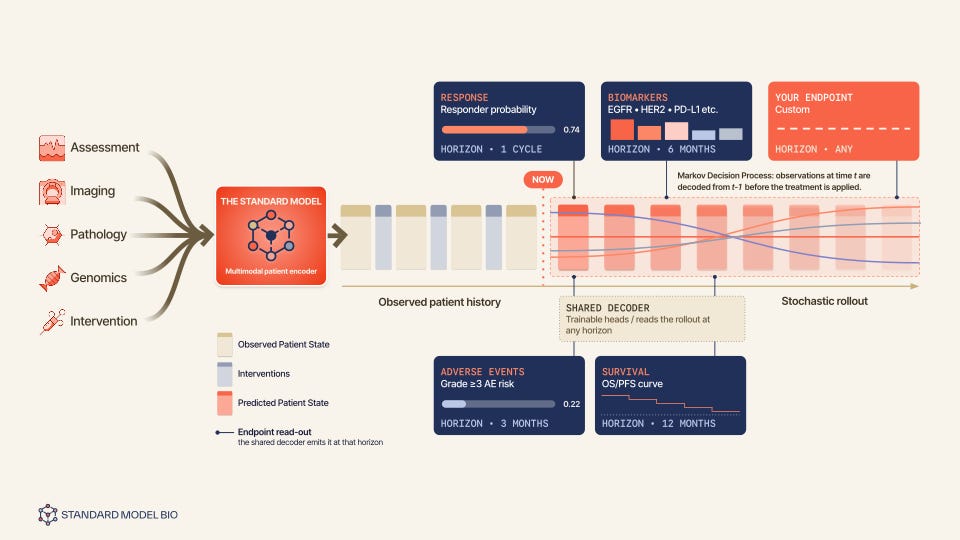
The architecture has three pieces.
An encoder takes whatever mix of genomics, pathology, imaging, clinical records, and past interventions happens to be available for a given patient, and fuses it into one fixed-size latent state at some reference time, with no assumption that every patient arrives with every modality.
A predictor, the actual simulator, then rolls that state forward in fixed time steps conditioned on whichever intervention is applied at each step: the next state is the current state, plus a drift shaped jointly by the state and the action, plus learned noise. Feed it the embedding for “give this drug at this dose” and it rolls forward the treated trajectory. Feed it a learned no-treatment token instead and the same drift function produces the untreated, natural-history trajectory. One model, one set of parameters, covers both counterfactual branches by construction, which is what makes it a simulator and not just a forecaster: you can swap the intervention and re-roll, the entire operation a counterfactual question requires.
A lightweight decoder then reads the simulated state and predicts only the handful of clinical observations/endpoints a real decision actually rides on: performance status, disease progression, biomarkers, adverse events, etc.
Early signals from the MSK CCDE oncology cohort
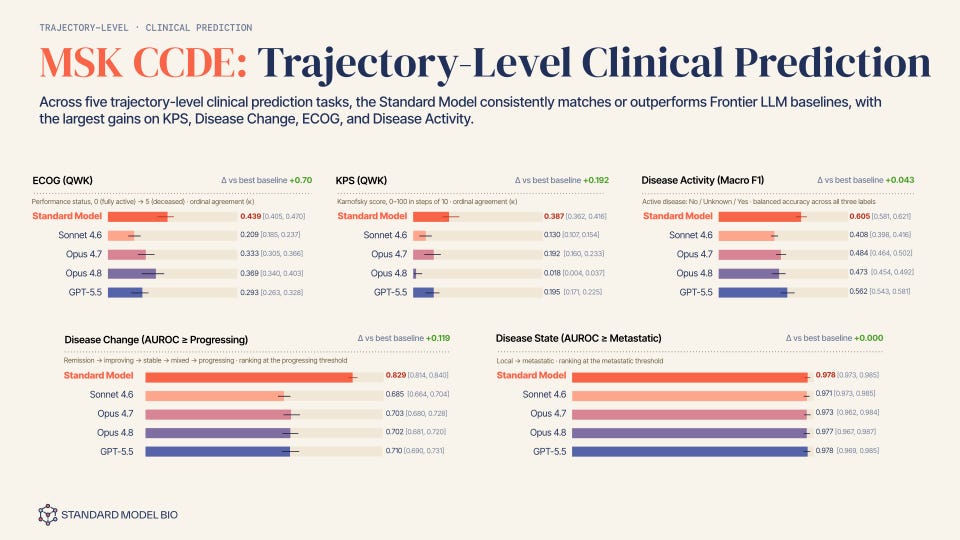
We ran the architecture against a real-world, longitudinal oncology cohort across 9 cancer types curated with Memorial Sloan Kettering, the MSK CCDE cohort, and the evaluation was built to match how the model is actually meant to be used. The model sees only a patient’s history (including gene mutation panel, pathology, imaging, other clinical assessments), up to the moment they started their first-line, index systemic cancer regimen, the point at which a clinician commits to a specific treatment and the counterfactual question actually becomes live. From that single starting state, the predictor rolls the trajectory forward in the model’s learned latent space, and the decoder reads five clinical endpoints off the rolled-out states, not off the input history itself. Two are ordinal and scored by quadratic weighted kappa (QWK) against the values clinicians actually recorded downstream: ECOG performance status, which runs from 0 (fully active) to 5 (deceased), and Karnofsky Performance Status. Two more are trajectory classifications scored by AUROC: whether the rolled-out disease course crosses a progressing threshold (remission through progressing) and whether it crosses from local to metastatic. The fifth predicts disease activity itself (active, inactive, or unknown) at that later point, scored on macro F1 across the three labels. Every one of the five is a genuine forward prediction: the model never sees the period it’s being asked to decode.
Disease change illustrates this perfectly, offering the sharpest version of the argument. The label isn’t “is this cancer bad.” It’s whether the specific regimen a clinician just started, on this specific patient, with this specific tumor biology and treatment history, is going to hold the disease in check or lose control of it over the following months. That’s not a fact sitting anywhere in the input. Nothing in the pre-regimen chart says whether the treatment will work; the chart only shows the state the treatment is about to act on. Answering it means simulating the drug-disease interaction itself, whatever mix of response, resistance, and decline actually plays out, and then reading a threshold off the far end of that simulated trajectory. A model that only pattern-matches the input state to a label has no way to get this right beyond the base rate. A model that rolls the state forward conditioned on the actual regimen given does.
Each of four current frontier general-purpose language models, Claude Sonnet 4.6, Opus 4.7, 4.8, and GPT-5.5, was given the same pre-index-regimen history, including rich, textualized multi-omic mutation panels, pathology and imaging findings to ensure strict data parity, and asked to predict the same downstream endpoints. In other words, LLMs see the exactly same inputs. These are, by a wide margin, the best general-purpose reasoners available off the shelf. If a purpose-built patient world model can’t beat them at rolling a trajectory forward from the same starting point, the argument in this piece is just a story.
StandardModel matched or outperformed the best baseline on all five tasks, and the size of the gap tracked the argument almost exactly: the largest, statistically clearest margins showed up on disease change, where the confidence intervals for StandardModel and the best baseline did not overlap at all.
The one task where the advantage disappeared was local-versus-metastatic disease state, where every model, ours included, landed within a point of 0.97 to 0.98 AUROC. That is the one endpoint on the list that is not really asking a model to roll anything forward: metastatic status largely determines which regimen a clinician picks in the first place, so by the time the encoder’s window closes it is closer to a fact already sitting in the history than an outcome the trajectory has to discover.
These are early, single-institution results, not a validated clinical claim, and they need to hold up across more data and more sites before treat them as settled. But they’re the first quantitative sign that an action-conditioned simulator, not simply a larger general-purpose model reading the same chart, is where the gains in patient-level prediction actually come from.
Isn’t this just a data and scale problem?
The real bottleneck for a patient world model isn’t the total volume of charts; it is the structural diversity of the treatments within them. To build an engine that doesn’t just hallucinate counterfactuals, the industry has to stop chasing massive, monolithic datasets that merely repeat the same clinical habits. Closing this gap requires a fundamentally different research program from the one currently attracting the most funding — not a scaled-up version of the same benchmarks, but multi-institution longitudinal cohorts and health system partnerships specifically chosen to expose identical biological baselines to completely different clinical choices. We don’t just need more data; we need evaluation designs that score forward prediction instead of state description, and far more variance in how the system is perturbed.
Problems not solved
The MSK CCDE numbers are preliminary and single-institution: evidence the architecture is worth pursuing, not proof the problem is solved. The real limits sit one level up, in the paradigm itself.
The deepest one: no patient is ever treated and left untreated on the same day to compare outcomes, so a simulator’s counterfactual branch, the whole point of building one, can never be checked directly against ground truth. It can only be checked against the realized branch and against existing randomized trials. The second: a patient’s chart, genomics, and imaging render their state, they are not the state itself. Adherence, unrecorded comorbidities, and social conditions shape outcomes but never enter that rendering, and no architecture fixes that without richer data. The third: regimens, doses, and combinations are sampled unevenly. Rolling a trajectory forward under an action the model has barely seen is extrapolation, and nothing guarantees the rollout stays plausible.
None of this kills the substrate of the approach; they define the roadmap. A model like this is not a substitute for a randomized trial; it narrows what is worth testing that way. Confidence in it should scale with scrutiny across more sites and comparisons, not one cohort’s numbers.
What to watch next
The next two years will be decided by a quiet realization: incremental improvements on retrospective observational benchmarks have become entirely non-causal. Squeezing an extra point of accuracy out of yesterday’s electronic health records usually just means a model has gotten better at exploiting data leakage, insurance billing cycles, or institutional habits, not that it actually understands human biology. Because these passive leaderboards are fundamentally saturated, the field will move toward a much more rigorous standard of proof.
Watch for a shift away from standard chart-matching and toward validating AI models against historical, retrospective randomized controlled trials. Because RCT data contains true, unconfounded causal ground truth, blinding a patient world model to these trials and forcing its individual simulated rollouts to validate if the model could forecast the treatment effects and survival curves is the ultimate litmus test. It proves whether a simulator can actually compute a true counterfactual or if it is merely memorizing clinical habits. Alongside that shift, watch for whether other groups working on patient-level trajectories converge on this same functional layout: an encoder for what is known, a predictor conditioned on the action taken, and a decoder for what a clinician actually needs to decide. If that pattern holds up under the strict causal scrutiny of historical trial data, AI for biomedicine may find its answer for the second half.
Subscribe to our Substack to be notified when publish new posts, or follow us on LinkedIn, Twitter/X and Huggingface for more.